

Hallmarks of Aging: Deep Dive
How a beautiful, self-healing mosaic of trillions of cells is slowly chipped away.

Motivation
First principle mechanics of aging have always been a topic that I poorly understood and given enormous progress in understanding reflected in scientific literature, there’s little excuse not to try to grok the issue at a molecular level. Over the next three posts I will explore causes of damage:
primary hallmarks
body’s response to damage: antagonistic hallmarks
culmination of damage: integrative hallmarks.
Most of the information here will be reflections and interpretations of a 2023 Cell review paper Hallmarks of aging: An expanding universe - Carlos López-Otín, Maria A. Blasco, Linda Partridge, Manuel Serrano, Guido Kroemer.
Introduction
It never ceases to amaze me how far the modern scientific method has gotten us. Diving into a review paper on a topic one is not an expert on already, typically results in multiple lateral deep-dives into topics explainign a new term or a fascinating side quest.
This time I ended up jotting down specific single atom interactions as they pertain to DNA oxidation, got side-tracked into history of a Sicilian cathedral and did a double take when I found out that blood cells are over 80% of total cell count in a human body.
Cells
A distributed system of microservices
We - humans - are not just vague cellular goo. There’s order to our organism. Neighbouring cells are likely to be the same tissue, form the same organ. Cells are spatially compartmentalised.
Each microservice (organ or cell) has a distinct role, operating independently yet contributing to the system's functionality. Strong API boundaries prevent unintended interactions. Biological barriers maintain order and prevent harmful crosstalk, such as inflammation. When a microservice overreaches or leaks data, it mirrors a failing biological barrier, causing system wide errors or chronic disease.
A mosaic
We’re a mosaic. A region of our body may have a set of cells that replicate from the same point mutation, creating a distinct tessera in a huge 3D mosaic of the human body.

Primary Hallmarks
What does hallmark mean in this aging scenario? It’s an indicator, perhaps a biomarker, perhaps a reflection of a biological process. And its change (or presence) is correlated with aging. It’s likely causal. But aging can also causally influence an indicator. An easy to grasp hallmark of aging is VO2 max for physiological tests.
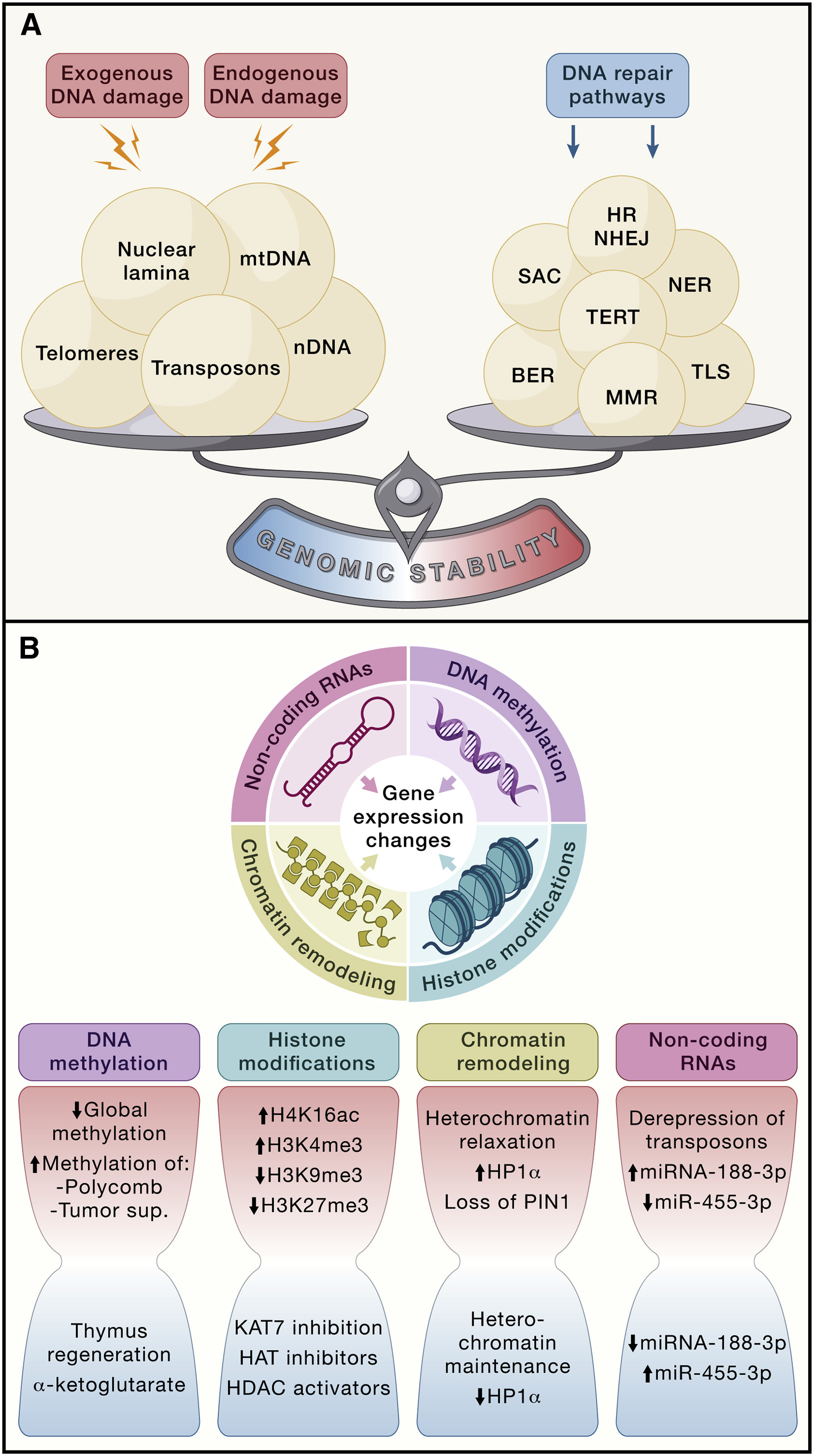
Review paper lists groups of damage categories and I will briefly go through them before focusing on a more holistic view of cell repair mechanisms. They are genomic instability, telomere attrition and epigenetic alterations.
Genomic Instability
Numbers game
The numbers game of DNA replication inside the cell’s nucleus makes me incredibly uncomfortable. The amount of mutations taking place on a regular basis that are caught by a series of mitigating processes makes aging feel dreadfully inevitable. The length of human DNA is about 3 billion base pairs. A one in a million mutation rate would lead to 3000 mutations in the cell.
For tissues that undergo a lot of environmental stress: esophagus tissue, for twenty year olds a few hundred mutations per cell are observed, going up to 2000 for 75 year olds. So even a tissue strongly exposed to the environment accumulated fewer than one in a million mutations over a human lifespan. Somatic mutant clones colonize the human esophagus with age, Science 2018.
Common mutations
Point mutation: C→T
Insertion: C→CCC
Deletion: CCC→C
Inversion: GATTACAGATTACA → GATTAAGACTTACA
Translocation: when sequences on two distinct chromosomes end up on a different chromosome during cell division.
Atomic level damage: oxygenation
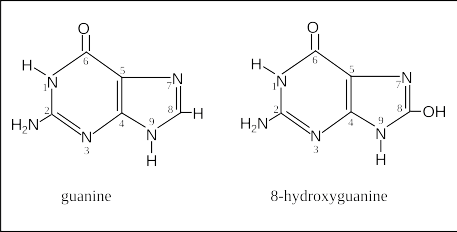
A very common type of damage to a DNA base is when it interacts with an oxygenating factors like -OH, O2, H2O2. These are byproducts of cellular metabolism. They are found mostly in mitochondria as that’s the cell’s powerhouse and some nasty reactive oxygen species are produced and emitted. A picture of a power plant spewing toxins into the air that land in a residential area. This particular process often leads to mutations as an oxygenated guanine can mispair with adenine (G:C→T:A) during replication.
{kind=link}
Atomic level damage: deamination
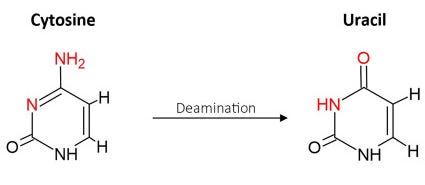
Guanine’s pair: adenine in turn can undergo deamination, which becomes uracil (a ribose that pairs with adenine again) leading to another G:C→T:A mutation. It can occur simply in contact with water and is more likely in acidic environments and higher temperatures also make the NH2 less stable.
Telomere Attrition
Why aren’t telomeres fully copied during division and replication? DNA polymerases - enzymes synthesising new DNA strands - need a specific starting point: the 3′-hydroxyl group. An -OH group attached to the third carbon atom of a sugar molecule. The 3' hydroxyl group acts as a reactive site, allowing new nucleotides to be added to a growing nucleic acid chain through the formation of a phosphodiester bond.

During DNA replication, the lagging strand cannot be fully synthesized at the chromosome ends due to the absence of a 3'-hydroxyl group for DNA polymerase to extend, leading to progressive chromosome shortening.

During DNA replication, the ends of chromosomes can't be fully copied because the machinery that builds DNA works like a zipper that needs a starter piece of fabric to get going. At the very end, there's no place to attach this starter piece, so a tiny bit of the DNA is left unzipped and lost each time. This problem is solved by an enzyme called telomerase which extends the parental strand with repetitive sequences using its RNA template, allowing the lagging strand to be completed.
See also below in mechanisms of DNA repair 7. TERT.
Epigenetic alterations
Methylation: Introduction
We already saw how individual atoms can cause a guanine and cytosine to be altered and be paired with thymine during replication. This is yet another mechanism by which cytosine is affected. However, cytosine methylation on its own is not mutagenic. A methylated cytosine is less likely to be expressed. So having the ability to silence gene expression via this single chemical process can be desirable.
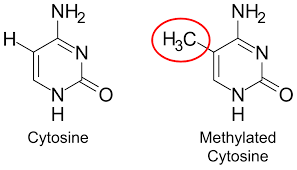
As a hallmark of aging, as organisms age the overall levels of DNA methylation across the genome decrease, this decrease is called global hypomethylation. As methylation is desirable in non-coding region, awakening them has negative consequences. Reduced methylation in promoter regions of DNA allows RNA polymerases easier access to DNA leading to increased gene expression.
Epigenetic Drift
One of the most intriguing mechanisms influencing the aging process is DNA methylation—a chemical modification where methyl groups attach to DNA and help regulate which genes are turned on or off. As we grow older, the carefully balanced patterns of methylation across our genome begin to shift. Scientists refer to this phenomenon as “epigenetic drift,” and it has profound consequences for cellular function, tissue maintenance, and our overall health. Understanding these changes provides valuable insight into why our bodies become more susceptible to diseases like cancer and neurodegeneration with age.
Global Hypomethylation
A key feature of aging is a decrease in global DNA methylation levels, often termed global hypomethylation. This drop in methylation primarily occurs in repetitive DNA sequences—such as LINEs, SINEs, and satellite DNA—along with various non-coding and intergenic regions of the genome. When these regions lose methylation, two major concerns arise. First, repetitive elements can become activated, potentially causing genomic instability and DNA damage. Second, genes that should remain silent may inadvertently switch on, fueling processes like unchecked cell growth and contributing to diseases such as cancer.
Targeted Hypermethylation
However, not all methylation changes in aging cells are decreases; some regions become more heavily methylated. Two important examples are the hypermethylation of Polycomb group (PcG) targets and tumour suppressor genes. PcG proteins typically control genes critical for development and cell differentiation, so when these regions become overly methylated, cells lose their ability to adapt and regenerate effectively. Meanwhile, hypermethylation of tumour suppressor genes like P16, BRCA1, and MLH1 silences these protective pathways. This leaves cells vulnerable to unregulated growth and mutation accumulation—factors that elevate the risk of cancer and other age-related diseases.
Aging Signature
Collectively, the decline in methylation across the genome, alongside localized increases in methylation at essential regulatory genes, forms a hallmark of aging. Global hypomethylation undermines genomic stability, while localised hypermethylation of critical genes compromises vital protective and regenerative pathways. Together, these alterations contribute to the broader epigenetic drift that disrupts normal gene regulation, ultimately driving many of the degenerative changes we associate with getting older.
Chromatin remodeling
What is chromatin? Chromatin is the complex of DNA and proteins that tightly winds and packs DNA strands. It’s not as tightly packed as when DNA is in a “chromosome” shape. The chromosome shape protects the insides of the DNA strands from transcription. The looser chromatin state makes DNA accessible for transcription, replication and repair. Visualization of DNA level processes will never cease to mesmerize.
Video 1. "Emerging Evidence of Chromosome Folding by Loop Extrusion". Fudenberg G, Abdennur N, Imakaev M, Goloborodko A, Mirny LA (2017). doi:10.1101/sqb.2017.82.034710 PMC 6512960. PMID 29728444.
So what’s with remodelling? Chromatin in the nucleus is supported by nuclear lamina - the inner wall of the nucleus. Defects in the lamina affect chromatin and by extension genomic stability.
Specific aging diseases, such as Hutchinson-Gilford Progeria Syndrome (HGPS) and Nestor-Guillermo Progeria Syndrome, are caused by mutations in two key proteins related to the nuclear lamina: LMNA and BANF1.
Mechanisms of DNA repair
In the paper a number of DNA repair processes are mentioned that affect genomic stability. Let’s go through them one by one.
1. BER (Base Excision Repair)
ELI5: A missing base on a double strand DNA. It has an abasic site. An enzyme sees what base is missing and fills it in.
Base Excision Repair (BER) is a precise mechanism that fixes small, non-helix-distorting damage to DNA bases, often caused by oxidation, alkylation, or deamination. It begins with a DNA glycosylase enzyme recognizing and removing the damaged base, leaving behind an abasic site. An enzyme called AP endonuclease then cleaves the DNA backbone at the abasic site. Next, DNA polymerase fills in the missing nucleotide using the undamaged complementary strand as a template, and DNA ligase seals the break to complete the repair. BER is crucial because these types of small damage events happen frequently due to normal cellular metabolism and environmental factors. Without BER, these small errors could accumulate, leading to mutations or genome instability. BER ensures that the DNA sequence is restored to its original, undamaged state, maintaining the fidelity of the genetic code and preventing diseases like cancer.
2. HR (Homologous Recombination)
ELI5: When there’s a break in both strands then we need a template to see which pair is missing. During cell division when all chromosomes are duplicated, it’s easy to do that HR based recombination.
Homologous Recombination (HR) is a high-fidelity DNA repair pathway that fixes double-strand breaks (DSBs) using an undamaged sister chromatid as a template. It is most active in the S and G2 phases of the cell cycle when a sister chromatid is available. The process begins with the recognition of the DSB and trimming of the broken ends by nucleases to create single-stranded DNA (ssDNA). These ssDNA regions are coated with RAD51, which facilitates the search for a homologous sequence on the sister chromatid. Once the homologous region is located, the damaged DNA strand uses it as a template to accurately repair the break through DNA synthesis and ligation. HR is crucial for maintaining genome stability because it ensures error-free repair. Defects in HR, such as in the BRCA1/2 genes, can lead to cancer, particularly breast and ovarian cancers.
3. NER (Nucleotide Excision Repair)
ELI5: Sometimes DNA “clumps”. Something as common as UV radiation can hit two adjacent T bases and cause them to form a covalent bond. The regular bond between thymine and adenine is a hydrogen bond. Hydrogen bonds work best along straight lines and are generally 10-20x weaker than covalent bonds. This weakness is a good thing. You don’t want your DNA to be difficult to unwind.
Read a detailed study of UV caused mutations. Formation of cyclobutane pyrimidine dimers at dipyrimidines containing 5-hydroxymethylcytosine Photochem Photobiol Sci, 2014.
Nucleotide Excision Repair (NER) removes bulky DNA lesions that distort the double helix, such as those caused by UV radiation (e.g., thymine dimers) or chemical damage. NER works by excising a short segment of the damaged DNA strand, followed by repair synthesis. The process starts when specific proteins, like XPC, recognize the distortion. Next, a multi-protein complex, including XPB and XPD helicases, unwinds the DNA around the lesion. Endonucleases like XPF and XPG cut the damaged strand on both sides of the lesion, removing a fragment of 24–32 nucleotides. DNA polymerase fills the gap using the undamaged strand as a template, and DNA ligase seals it. NER is essential for preventing mutations and diseases like xeroderma pigmentosum (XP), where individuals are hypersensitive to UV light due to defective NER, leading to high rates of skin cancer.
4. NHEJ (Non-Homologous End Joining)
ELI5: When there’s no template but both strands are severed, the unequal dangly bits are chopped off and DNA is simply glued together. It’s error prone of course. But presumably a win over a total seizure of replication.
Non-Homologous End Joining (NHEJ) is a rapid, error-prone repair mechanism that fixes double-strand breaks (DSBs) without requiring a homologous template. It operates throughout the cell cycle but is especially critical during the G1 phase, when a sister chromatid is unavailable. In NHEJ, the broken DNA ends are recognized and bound by the Ku70/Ku80 protein complex, which recruits a DNA-PKcs kinase to stabilize the ends. If the ends are not perfectly compatible, nucleases may trim them, and polymerases can add random nucleotides to allow ligation. Finally, DNA ligase IV, along with its cofactor XRCC4, seals the break. While NHEJ is efficient, its error-prone nature can lead to small insertions or deletions (indels) at the repair site. Despite this, NHEJ is essential for maintaining genome integrity in non-dividing cells and plays a key role in immune system development through V(D)J recombination.
5. MMR (Mismatch Repair)
ELI5: Post replication stage repair of errors of insertion or deletion. Again, based on the paired base (C-G, A-T).
Mismatch Repair (MMR) is a post-replication repair system that corrects base-pair mismatches and small insertions or deletions introduced during DNA replication. These errors occur when DNA polymerase incorporates the wrong nucleotide or slips on repetitive sequences. The process begins with MutS proteins recognizing the mismatch and binding to it. Then, MutL scans the DNA to locate a nick on the newly synthesized strand, marking it as the one to repair. Exonucleases remove the mismatched section, and DNA polymerase fills the gap using the parent strand as a template. DNA ligase seals the repair. MMR is critical for maintaining replication fidelity, and defects in MMR proteins, such as MLH1 and MSH2, are linked to Lynch syndrome, a hereditary cancer predisposition syndrome that increases the risk of colorectal and other cancers.
7. TERT (Telomerase Reverse Transcriptase)
ELI5: TERT is rebuilding and extending the lengths of telomers following division and replication.
Telomerase Reverse Transcriptase (TERT) is an enzyme that maintains the length of telomeres, repetitive DNA sequences at the ends of chromosomes. Telomeres protect chromosome ends from being recognized as DNA breaks. During replication, telomeres shorten because DNA polymerase cannot fully replicate the ends of linear DNA (the "end replication problem"). TERT, the catalytic subunit of telomerase, extends the telomeres by adding repeats (e.g., TTAGGG in humans) using an RNA template within the telomerase complex. Telomerase is active in stem cells, germ cells, and cancer cells but is generally inactive in somatic cells. Telomere shortening contributes to cellular senescence and aging. Reactivating telomerase could theoretically delay aging, but excessive activity is a hallmark of many cancers, highlighting the need for balance in its regulation.
8. TLS (Trans-Lesion Synthesis)
ELI5: TLS is the last line of defense for error correction during copying: when there’s a missing base or the two TTs which created a covalent bond are found, there are additional helpers that inject a random base. There’s no error correction and so it’s just a solution that’s “better” than completely stopping replication.
Trans-Lesion Synthesis (TLS) is a DNA damage tolerance mechanism that allows cells to bypass lesions that block normal DNA replication, such as thymine dimers or abasic sites. When the replication fork stalls at a lesion, specialized low-fidelity polymerases, like Pol eta (η) or Pol kappa (κ), are recruited to synthesize DNA across the damage. These polymerases are error-prone because they lack proofreading activity, so TLS often introduces mutations at the lesion site. However, TLS prevents replication fork collapse, which could lead to double-strand breaks or genome instability. TLS is particularly important for survival under conditions of high DNA damage, such as during UV exposure. Mutations in TLS components, like Pol η, cause diseases such as xeroderma pigmentosum variant (XP-V), which leads to UV sensitivity and skin cancer susceptibility.
RNA therapies
Emerging RNA therapies such as antisense oligonucleotides (ASOs) offer promising treatments of lamina and chromatin remodeling.
In HGPS, the aberrant protein progerin, resulting from a splicing defect in the LMNA gene, disrupts nuclear lamina integrity and accelerates cellular aging. ASO therapies target the defective mRNA responsible for progerin production. By blocking the abnormal splicing or degrading the faulty mRNA, ASOs can reduce progerin levels and alleviate disease symptoms, offering a hopeful strategy for treating HGPS.
Although NGPS results from mutations in BANF1, similar RNA-based therapeutic approaches could be developed to address the underlying molecular defects. For example, ASOs or other RNA therapies might correct splicing errors, restore normal protein function, or mitigate downstream effects of the mutation.
Antisense oligonucleotides (ASOs) are designed to specifically bind to the mRNA responsible for producing progerin through complementary base pairing, similar to the way the two strands of DNA interact.
By attaching to the target mRNA, ASOs prevent the ribosome from reading it, effectively halting the translation of progerin protein.
This approach is classified as RNA-based therapy or RNA-targeted therapy, as it works at the RNA level and is temporary in nature. It is not considered "gene therapy," which involves permanent changes to the DNA.